In het zelfstudie document hebben we het gehad over dichotome uitkomstvariabelen en over statistische analyse van tabellen. Vraag 1 laat je oefenen met het berekenen van de juiste associatiematen en het uitvoeren van de chi-kwadraat toets op medische data. Vraag 2 geeft een statistische kijk op het artikel over genome-wide association en vraag 3 laat jullie kennis maken met mogelijke verstorende variabelen, waarna we wat uitgebreider ingaan op het gebruik van R en de valkuilen die mogelijk optreden.
Stappen van data analyse
We gebruiken steeds de stappen van data analyse zoals beschreven in PSLS:
State. Wat is de onderzoeksvraag? Geef de praktische context erbij.
Plan. Welke specifieke statistische handelingen/berekeningen hebben we nodig? Wanneer we toetsen, noemen we hier expliciet de nul- en alternatieve hypotheses.
Solve. Gebruik geschikte visualisaties en/of berekeningen om de vraag te beantwoorden. Bij het toetsen (met betrouwbaarheidsinterval of p-waarde), checken we eerst (zo mogelijk) de aannames voor inferentie.
Conclude. Geef je conclusie in de context van de onderzoeksvraag.
Vraag 1
Uit een groot observationeel onderzoek (het ERGO onderzoek) onder 55-jarige mannen en vrouwen is het verband onderzocht tussen roken en het optreden van hart- en vaatziekten (HVZ). 7685 personen zijn geïncludeerd en vervolgens gedurende 5 jaar gevolgd. Hieronder zijn de gegevens in tabelvorm weergegeven.
| Rookt nu | HVZ ja | HVZ nee | Totaal |
|---|---|---|---|
| Ja | 41 | 1695 | 1736 |
| Nee | 107 | 5842 | 5949 |
| Totaal | 148 | 7537 | 7685 |
a. Welke associatiematen kunnen we hier berekenen?
Prospectief cohortonderzoek, dus we mogen RD, RR en OR gebruiken.
b. Gebruik de rekenmachine om deze associatiematen te berekenen en leg in woorden uit wat ze betekenen.
We willen het al of niet verhoogde risico op HVZ weten van roken t.o.v. niet roken. Het risico voor rokers is 41/1736 = 0.024 en voor niet-rokers 107/5949 = 0.018.
Het verschil (RD) is dus 0.006 ofwel 0.6%. Dit is een positief getal, dus dit representeert een hoger risico op HVZ voor rokers t.o.v. niet-rokers.
Het Relatieve Risico (RR) is 0.024 / 0.018 = 1.33. Dit is groter dan 1, dus er is een hoger risico op HVZ voor rokers t.o.v. niet-rokers.
De OR = (41/1695) / (107/5842) = 1.32. Ook de OR is groter dan 1, dus de odds op HVZ is groter voor rokers t.o.v. niet-rokers.
Omdat de prevalentie van HVZ heel laag is (1.9%) lijken de RR en OR hier heel goed op elkaar.
c. Gebruik het vier stappenplan om deze data te toetsen. Gebruik R voor je data-analyse.
# Data invoeren
roken_hvz <- matrix(c(41, 1695, 107, 5842), nrow = 2, byrow = TRUE)
colnames(roken_hvz) <- c("HVZ Ja", "HVZ Nee")
rownames(roken_hvz) <- c("Rookt Ja", "Rookt Nee")
roken_hvz## HVZ Ja HVZ Nee
## Rookt Ja 41 1695
## Rookt Nee 107 5842# Proporties berekenen
prop.table(roken_hvz, 1)## HVZ Ja HVZ Nee
## Rookt Ja 0.02361751 0.9763825
## Rookt Nee 0.01798622 0.9820138# Chi-kwadraat toets
chisq.test(roken_hvz, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: roken_hvz
## X-squared = 2.2563, df = 1, p-value = 0.1331State. Is er een verband tussen roken en Hart- en Vaatziekten?
Plan. H0: er is geen verband, H1: er is wel een verband. Zowel de determinant als de uitkomst zijn dichotome variabelen, dus gebruiken we hier een chi-kwadraat toets met 1 vrijheidsgraad (df). We hanteren de gebruikelijke alfa 0.05.
We checken de aanname dat de verwachte waarden in iedere cel groter dan 5 moet zijn (80% van 4 is 3.2, dus bij een 2x2 tabel moeten alle cellen 5 of meer observaties hebben). Hier wordt aan voldaan.
Solve. Proporties, RD, RR en OR staan hierboven aangegeven. Dit kun je eventueel nog in een staafdiagram visualiseren, maar de getallen spreken voor zich. Om de chi-kwadraat toets uit te voeren gebruik je:
chisq.test(rbind(c(41,1695),c(107,5842)), correct = FALSE)##
## Pearson's Chi-squared test
##
## data: rbind(c(41, 1695), c(107, 5842))
## X-squared = 2.2563, df = 1, p-value = 0.1331Dit resulteert in een X-squared = 2.2563, df = 1, p-value = 0.1331
Conclude. We verwerpen de nul-hypothese dus niet, en concluderen dat er niet voldoende bewijs is voor een verband tussen roken en HVZ.
Vraag 2
In deze opgave zullen jullie het artikel “a genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults” van Perez-Andreu et al. bekijken vanuit het statistisch perspectief.
a. Wat is de incidentie en/of prevalentie van ALL? Probeer dit op te zoeken op het internet. Kunnen we dit ook bepalen aan de hand van de data in dit artikel? Hoe? Of waarom niet?
Antwoord is niet snel te vinden en nogal divers. Hangt af van leeftijd en geografische locatie. Onderzoek is veelal gericht op mensen in de verenigde staten. https://seer.cancer.gov/statfacts/html/alyl.html geeft een duidelijk overzicht van de nieuwe casussen per jaar (incidentie) over de jaren heen en splitst het uit per bevolkingsgroepen.
Nee, dit kunnen we niet bepalen. Het is een case control onderzoek. Let op! De controles zijn uit andere studies. Deze deelnemers hebben geen ALL maar potentieel atherosclerose (discovery sample), Aids, astma, schizofrenie, bipolaire stoornis (replication sample). Uit de beschrijving is niet duidelijk of alleen controles uit die studies zijn geselecteerd of iedereen.
b. Hoeveel cases en controles zitten er in de discovery en replication studie? Check deze aantallen in tabel 1.
In de tekst: Discovery sample: 308 cases en 6661 controles, Replication sample: 162 cases en 5755 controles. Aantallen in de tabel zijn licht afwijkend: 306 (SNP1) / 307 cases (SNP2) in discovery sample en 161 (SNP1) / 157 cases (SNP2) in replication sample. Wordt ergens in het artikel vermeld waarom dit is? De afwijking is erg klein, dus we hoeven ons geen zorgen te maken om selectieve uitval. 6661,6659,5753/5747 voor controles).
c. In het artikel worden 2 SNP’s gepresenteerd (uit een set van 635297 SNP’s) die significant samenhangen met ALL in de AYA group. In tabel 1 (hieronder nogmaals weergegeven) staan de gegevens van deze SNP’s samengevat.
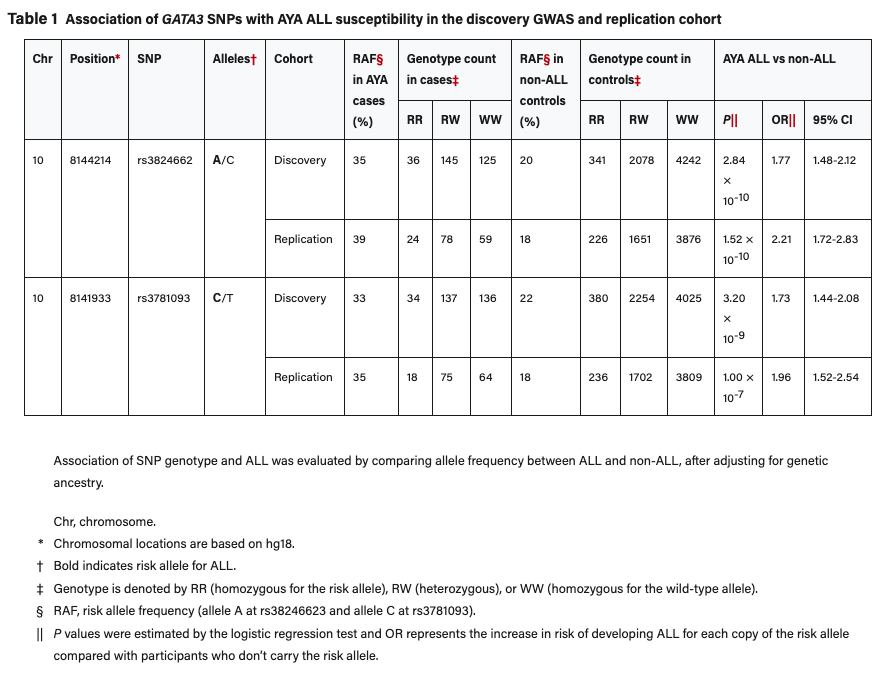
De belangrijkste vraag is of het hebben van een risico-allel de odds op het krijgen van ALL beïnvloedt. De onderzoekers hebben dit met een GWAS voor vele SNP’s onderzocht. Wij zullen op een simpelere manier de analyses voor de twee succesvolste SNP’s uit deze studie herhalen. We maken daarbij in eerste instantie de assumptie dat het risico-allel dominant is. Dat wil zeggen dat het homozygote genotype met twee risico-allellen een even grote verandering in risico op ALL heeft als het heterozygote genotype (de carrier) ten opzichte van het homozygote “wild” genotype. Om die reden kunnen we de eerste twee groepen samenvoegen.
Gebruik de gegevens in de tabel om voor beide cohorten (discovery en replication) en voor beide SNP’s een 2x2 tabel en een 2x3 tabel op te stellen. Beantwoord daarna de volgende vragen.
d. Wat zijn de odds ratios op ALL voor personen met risicodragend allel t.o.v. personen zonder risicodragend allel voor beide SNP’s en voor beide cohorten? Komt dit overeen met de gerapporteerde odds ratios in tabel 1 van het artikel?
# rs3824662 - discovery
SNP1.disc <- rbind(c(36+145, 341+2078), c(125, 4242))
colnames(SNP1.disc) <- c("Cases", "Controls")
rownames(SNP1.disc) <- c("RR+RW", "WW")
SNP1.disc## Cases Controls
## RR+RW 181 2419
## WW 125 4242# rs3824662 - replication
SNP1.rep <- rbind(c(24+78, 226+1651), c(59, 3876))
colnames(SNP1.rep) <- c("Cases", "Controls")
rownames(SNP1.rep) <- c("RR+RW", "WW")
SNP1.rep## Cases Controls
## RR+RW 102 1877
## WW 59 3876# rs3781093 - discovery
SNP2.disc <- rbind(c(34+137, 380+2254), c(136, 4025))
colnames(SNP2.disc) <- c("Cases", "Controls")
rownames(SNP2.disc) <- c("RR+RW", "WW")
SNP2.disc## Cases Controls
## RR+RW 171 2634
## WW 136 4025# rs3781093 - replication
SNP2.rep <- rbind(c(18+75, 236+1702), c(64, 3809))
colnames(SNP2.rep) <- c("Cases", "Controls")
rownames(SNP2.rep) <- c("RR+RW", "WW")
SNP2.rep## Cases Controls
## RR+RW 93 1938
## WW 64 3809# rs3824662 - discovery
epi.2by2(dat = SNP1.disc, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 181 2419 2600 6.96 (6.01 to 8.01)
## Exposure- 125 4242 4367 2.86 (2.39 to 3.40)
## Total 306 6661 6967 4.39 (3.92 to 4.90)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 2.43 (1.95, 3.04)
## Inc odds ratio 2.54 (2.01, 3.21)
## Attrib risk in the exposed * 4.10 (3.00, 5.20)
## Attrib fraction in the exposed (%) 58.88 (48.65, 67.08)
## Attrib risk in the population * 1.53 (0.84, 2.22)
## Attrib fraction in the population (%) 34.83 (30.59, 39.12)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 65.213 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population units# rs3824662 - replication
epi.2by2(dat = SNP1.rep, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 102 1877 1979 5.15 (4.22 to 6.22)
## Exposure- 59 3876 3935 1.50 (1.14 to 1.93)
## Total 161 5753 5914 2.72 (2.32 to 3.17)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 3.44 (2.51, 4.72)
## Inc odds ratio 3.57 (2.58, 4.94)
## Attrib risk in the exposed * 3.65 (2.61, 4.70)
## Attrib fraction in the exposed (%) 70.91 (60.15, 78.76)
## Attrib risk in the population * 1.22 (0.66, 1.79)
## Attrib fraction in the population (%) 44.92 (39.11, 50.78)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 66.415 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population units# rs3781093 - discovery
epi.2by2(dat = SNP2.disc, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 171 2634 2805 6.10 (5.24 to 7.05)
## Exposure- 136 4025 4161 3.27 (2.75 to 3.85)
## Total 307 6659 6966 4.41 (3.94 to 4.92)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 1.87 (1.50, 2.32)
## Inc odds ratio 1.92 (1.53, 2.42)
## Attrib risk in the exposed * 2.83 (1.79, 3.87)
## Attrib fraction in the exposed (%) 46.39 (33.22, 56.96)
## Attrib risk in the population * 1.14 (0.41, 1.86)
## Attrib fraction in the population (%) 25.84 (21.59, 30.17)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 31.803 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population units# rs3781093 - replication
epi.2by2(dat = SNP2.rep, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 93 1938 2031 4.58 (3.71 to 5.58)
## Exposure- 64 3809 3873 1.65 (1.27 to 2.11)
## Total 157 5747 5904 2.66 (2.26 to 3.10)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 2.77 (2.02, 3.79)
## Inc odds ratio 2.86 (2.07, 3.94)
## Attrib risk in the exposed * 2.93 (1.93, 3.92)
## Attrib fraction in the exposed (%) 63.91 (50.67, 73.60)
## Attrib risk in the population * 1.01 (0.43, 1.58)
## Attrib fraction in the population (%) 37.86 (32.13, 43.69)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 44.084 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population unitsHet is handig om hier de 2x2 tabellen weer op te stellen voordat je de analyses gaat doen:
rs3824662 – discovery
| Genotype | AYA-ALL (cases) | Non-ALL (controles) |
|---|---|---|
| RR+RW | 181 | 2419 |
| WW | 125 | 4242 |
rs3824662 – replication
| Genotype | AYA-ALL (cases) | Non-ALL (controles) |
|---|---|---|
| RR+RW | 102 | 1877 |
| WW | 59 | 3876 |
rs3781093 – discovery
| Genotype | AYA-ALL (cases) | Non-ALL (controles) |
|---|---|---|
| RR+RW | 171 | 2634 |
| WW | 136 | 4025 |
rs3781093 – replication
| Genotype | AYA-ALL (cases) | Non-ALL (controles) |
|---|---|---|
| RR+RW | 93 | 1938 |
| WW | 64 | 3809 |
Berekeningen:
- rs3824662 – discovery OR = (181/(2419))/(125/(4242)) = 2.5392
- rs3824662 – replication OR = (102/(59))/(1877/(3876)) = 3.5699
- rs3781093 – discovery OR = (171/(136))/(2634/(4025)) = 1.921354
- rs3781093 – replication OR = (93/(64))/(1938/(3809)) = 2.8560
Deze OR’s, berekend op de 2x2 tabellen, zijn hoger dan de OR’s in de tabel, die gebaseerd zijn op een additief model in plaats van een dominant model (zie de laatste opmerking bij de tabel).
e. Interpreteer de OR’s en hun betrouwbaarheidsintervallen in tabel 1 van het artikel.
De OR’s in het artikel hebben allemaal een waarde groter dan 1, en een CI dat 1 niet bevat. (Dit is natuurlijk niet gek in een tabel waar de succesvolle SNP’s worden gepresenteerd). De ware populatie OR ligt met 95% zekerheid tussen de grenzen van de betrouwbaarheidsintervallen. Omdat deze studie zowel een discovery als een replicatie sample bevat hebben we nu 2 BHI’s waar met 95% zekerheid de ware OR in zal zitten. Dit sterkt ons natuurlijk enorm in de conclusie van deze effecten: mensen die het risico allel dragen bij de twee SNP’s hebben verhoogde odds op ALL. Deze verhoogde odds kwamen naar boven bij zowel de discovery als de replicatie cohorts.
f. Formuleer de nulhypothese en de alternatieve hypothese voor de 2x2 tabellen.
H0: er is geen verband tussen genotype en ziekte (of: OR=1)
H1: er is wel verband tussen genotype en ziekte (of: OR≠1)
We toetsen met een type I fout van 0.05. Omdat er meerdere toetsen zijn uitgevoerd is er een correctie voor de kritieke alfa. Alleen p-waardes kleiner dan 5*10^-8 zijn significant
g. Check de aannames van een chi-kwadraat toets voordat je deze uitvoert.
Een goede manier om dit te doen is eerst te kijken welke cel de kleinste verwachte waarde zal hebben. Dit is de cel die hoort bij de laagste waarde van de rij-totalen en de laagste waarde van de kolomtotalen. Als deze waarde groter dan 5 is hoef je niet meer verder te zoeken, want de rest is dan in ieder geval nog groter. In dit geval kijken we dus naar de formule (rijtotaalkolomtotaal)/tabeltotaal. Bij SNP1 van het disc sample is dit de cel met outcome+ en exposed+. We rekenen dan: (2600306)/6967 = 114,20. Dit is veel groter dan 5, dus aan de voorwaarde is voldaan. De voorwaarde van onafhankelijke cases bestaat natuurlijk ook, maar die kunnen we niet checken met getallen.
h. Voer voor beide cohorten en beide SNP’s de Pearson’s chi-kwadraat toets uit in R.
# Chi-kwadraat toetsen
chisq.test(SNP1.disc, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP1.disc
## X-squared = 65.213, df = 1, p-value = 0.0000000000000006724chisq.test(SNP1.rep, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP1.rep
## X-squared = 66.415, df = 1, p-value = 0.0000000000000003653chisq.test(SNP2.disc, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP2.disc
## X-squared = 31.803, df = 1, p-value = 0.00000001706chisq.test(SNP2.rep, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP2.rep
## X-squared = 44.084, df = 1, p-value = 0.00000000003146Voor SNP1.disc geldt: chi2(1) = 65.213, p-waarde <0.001
Voor SNP1.rep geldt: chi2(1) = 66.415, p-waarde <0.001
Voor SNP2.disc geldt: chi2(1) = 31.803, p-waarde <0.001
Voor SNP2.rep geldt: chi2(1) = 44.084, p-waarde <0.001
i. Herhaal deze stappen voor de 2x3 tabellen voor beide cohorten en beide SNP’s.
# 2x3 tabellen maken
SNP1.disc.2x3 <- rbind(c(36, 341), c(145, 2078), c(125, 4242))
colnames(SNP1.disc.2x3) <- c("Cases", "Controls")
rownames(SNP1.disc.2x3) <- c("RR", "RW", "WW")
SNP1.disc.2x3## Cases Controls
## RR 36 341
## RW 145 2078
## WW 125 4242SNP1.rep.2x3 <- rbind(c(24, 226), c(78, 1651), c(59, 3876))
colnames(SNP1.rep.2x3) <- c("Cases", "Controls")
rownames(SNP1.rep.2x3) <- c("RR", "RW", "WW")
SNP1.rep.2x3## Cases Controls
## RR 24 226
## RW 78 1651
## WW 59 3876SNP2.disc.2x3 <- rbind(c(34, 380), c(137, 2254), c(136, 4025))
colnames(SNP2.disc.2x3) <- c("Cases", "Controls")
rownames(SNP2.disc.2x3) <- c("RR", "RW", "WW")
SNP2.disc.2x3## Cases Controls
## RR 34 380
## RW 137 2254
## WW 136 4025SNP2.rep.2x3 <- rbind(c(18, 236), c(75, 1702), c(64, 3809))
colnames(SNP2.rep.2x3) <- c("Cases", "Controls")
rownames(SNP2.rep.2x3) <- c("RR", "RW", "WW")
SNP2.rep.2x3## Cases Controls
## RR 18 236
## RW 75 1702
## WW 64 3809# Chi-kwadraat toetsen op 2x3 tabellen
chisq.test(SNP1.disc.2x3, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP1.disc.2x3
## X-squared = 72.243, df = 2, p-value < 0.00000000000000022chisq.test(SNP1.rep.2x3, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP1.rep.2x3
## X-squared = 87.773, df = 2, p-value < 0.00000000000000022chisq.test(SNP2.disc.2x3, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP2.disc.2x3
## X-squared = 36.966, df = 2, p-value = 0.000000009394chisq.test(SNP2.rep.2x3, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: SNP2.rep.2x3
## X-squared = 51.136, df = 2, p-value = 0.000000000007871Hypotheses (blijven hetzelfde):
H0: er is geen verband tussen genotype en ziekte
H1: er is wel verband tussen genotype en ziekte
We toetsen met een type I fout van 0.05. Omdat er meerdere toetsen zijn uitgevoerd is er een correctie voor de kritieke alfa. Alleen p-waardes kleiner dan 5*10^-8 zijn significant
2 bij 3 tabellen, test uitgevoerd met chisq.test(SNP1.disc.2x3, correct = F):
Voor SNP1.disc geldt: chi2(2) = 72.243, p-waarde <0.001
Voor SNP1.rep geldt: chi2(2) = 87.773, p-waarde <0.001
Voor SNP2.disc geldt: chi2(2) = 36.966, p-waarde <0.001
Voor SNP2.rep geldt: chi2(2) = 51.136, p-waarde <0.001
j. Welke assumptie hebben we nu losgelaten? Hoe moet je de resultaten van de chi-kwadraat toets nu interpreteren?
We laten hier de assumptie van dominantie los. Het kan dus ook het additieve model zijn, zoals in het artikel gebruikt is. En in theorie zou het zelfs zo kunnen zijn dat alleen de heterozygote vorm afwijkt van de 2 homozygote vormen. Het enige dat we testen is of de verhoudingen in minimaal 1 van deze genotypes afwijkt van de andere.
Vraag 3
Om de effectiviteit van een bepaalde therapie te onderzoeken worden 1000 mensen met een ernstige aandoening wel (T+) of niet (T-) met de therapie behandeld. Na een vaste tijd wordt gekeken of de patiënt overleden is (O+) of niet (O-).
De resultaten staan in onderstaande kruistabel samengevat:
Mannen
| O+ | O- | Totaal | |
|---|---|---|---|
| T+ | 120 | 280 | 400 |
| T- | 80 | 20 | 100 |
| Totaal | 200 | 300 | 500 |
Vrouwen
| O+ | O- | Totaal | |
|---|---|---|---|
| T+ | 3 | 97 | 100 |
| T- | 32 | 368 | 400 |
| Totaal | 35 | 465 | 500 |
a. Bereken (met rekenmachine) het relatieve risico (RR) op overlijden (O+) voor mannen die de therapie wel hebben gehad (T+) tegen de mannen die hem niet hebben gehad (T-). Hebben mannen die de therapie hebben gevolgd een grotere of kleinere kans op overlijden? (je hoeft hier nog geen rekening te houden met steekproefvariatie).
RRman = (120/400) / (80/100) = 0.375
Er is een kleinere kans op overlijden als de mannen therapie hebben gehad (want RR<1).
b. Bepaal tevens de odds ratio (OR). Wat valt je op?
ORman = (120/280) / (80/20) = 0.107
OR en RR wijken erg van elkaar af.
c. Voer stap a en b ook uit voor de vrouwen.
RRvrouw = (3/100) / (32/400) = 0.375
ORvrouw = (3/97) / (32/368) = 0.356
d. Waarom wijken OR en RR bij mannen zoveel af, en bij vrouwen niet?
De incidentie bij mannen is hoog. 40% van alle mannen overlijdt. De incidentie bij vrouwen is juist erg laag. Slechts 7% van de vrouwen overlijdt.
e. Voeg beide datasets bij elkaar en bereken nogmaals OR. Wat valt je op aan de berekende OR van de hele groep ten opzichte van de twee oorspronkelijke groepen? Hoe komt dat?
Gehele groep
| Therapie | O+ | O- | Totaal |
|---|---|---|---|
| T+ | 123 | 377 | 500 |
| T- | 112 | 388 | 500 |
| Totaal | 235 | 765 | 1000 |
ORallen = (123/377) / (112/388) = 1.130
Dit is een heel andere waarde. Sterker nog: de OR is groter dan 1, en dat zou betekenen dat er een hoger risico is op overlijden als je de therapie volgt! In plaats van een beschermend effect heeft het hier een versterkend effect.
Dit is een voorbeeld van confounding. De relatie tussen therapie en overlijden is in deze observationele studie verstoord door het geslacht van de deelnemers. Er spelen hier twee effecten een rol. Over het algemeen overlijden er meer mannen dan vrouwen. En tegelijkertijd hebben veel mannen therapie gekregen (400 van de 500) terwijl de meerderheid van de vrouwen geen therapie kreeg (slechts 100 van de 500 vrouwen kregen therapie). Dit betekent dat de groep die therapie kreeg voornamelijk uit mannen bestond, terwijl de andere groep voornamelijk uit vrouwen bestond. Maar deze personen hebben dus over het algemeen al een groot verschil in kans op overlijden. Het werkelijke effect van therapie is daardoor niet te zien als je de analyse op de hele groep uitvoert. We moeten deze analyse dus voor beide groepen apart uitvoeren. Er zijn overigens wel mogelijkheden om dit in een analyse te doen (o.a. logistische regressie).
f. Voer de data-analyse voor mannen en vrouwen apart ook uit met R zoals je in de zelfstudie hebt geleerd, en interpreteer de resultaten.
# Data voor mannen
man.tab <- rbind(c(120, 280), c(80, 20))
colnames(man.tab) <- c("O+", "O-")
rownames(man.tab) <- c("T+", "T-")
man.tab## O+ O-
## T+ 120 280
## T- 80 20# Chi-kwadraat toets voor mannen
chisq.test(man.tab, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: man.tab
## X-squared = 83.333, df = 1, p-value < 0.00000000000000022# Data voor vrouwen
vrouw.tab <- rbind(c(3, 97), c(32, 368))
colnames(vrouw.tab) <- c("O+", "O-")
rownames(vrouw.tab) <- c("T+", "T-")
vrouw.tab## O+ O-
## T+ 3 97
## T- 32 368# Chi-kwadraat toets voor vrouwen
chisq.test(vrouw.tab, correct = FALSE)##
## Pearson's Chi-squared test
##
## data: vrouw.tab
## X-squared = 3.0722, df = 1, p-value = 0.07964# Proporties
prop.table(man.tab, 1)## O+ O-
## T+ 0.3 0.7
## T- 0.8 0.2prop.table(vrouw.tab, 1)## O+ O-
## T+ 0.03 0.97
## T- 0.08 0.92Voor mannen:
X-squared = 83.333, df = 1, p-value < 2.2e-16
Voor vrouwen:
X-squared = 3.0722, df = 1, p-value = 0.07964
We zien hier dat het verband bij mannen wel significant is en bij vrouwen niet.
g. Meestal zullen we data in een datafile hebben, en laten we R een tabel genereren die we vervolgens analyseren. Dit kunnen we hier ook oefenen. Open en bekijk daarvoor de volgende datafile: therapieoverlijden.csv (vergeet niet je eigen foldernaam aan te passen!):
# Let op: pas het pad aan naar waar je datafile staat
# Indir <- "O:/Biostatistiek/Onderwijs/Biomedische_wetenschappen/Bioinformatica/2021-2022/leereenheid 4 - categorische uitkomsten/"
# d <- data.frame(read.table(file.path(Indir, "therapieoverlijden.csv"), sep=",", header = TRUE))
# head(d)
# Voor nu maken we de data handmatig aan
d <- data.frame(
geslacht = c(rep("man", 500), rep("vrouw", 500)),
therapie = c(rep("T+", 400), rep("T-", 100), rep("T+", 100), rep("T-", 400)),
overlijden = c(rep("O+", 120), rep("O-", 280), rep("O+", 80), rep("O-", 20),
rep("O+", 3), rep("O-", 97), rep("O+", 32), rep("O-", 368))
)
head(d)## geslacht therapie overlijden
## 1 man T+ O+
## 2 man T+ O+
## 3 man T+ O+
## 4 man T+ O+
## 5 man T+ O+
## 6 man T+ O+# Tabel maken voor alle observaties
th.ov.tab <- table(d$therapie, d$overlijden, dnn = c("therapie", "overlijden"))
th.ov.tab## overlijden
## therapie O- O+
## T- 388 112
## T+ 377 123prop.table(th.ov.tab, 1)## overlijden
## therapie O- O+
## T- 0.776 0.224
## T+ 0.754 0.246# Selecteren van mannen
d.man <- d[d$geslacht == "man",]
man.th.ov.tab <- table(d.man$therapie, d.man$overlijden, dnn = c("therapie", "overlijden"))
man.th.ov.tab## overlijden
## therapie O- O+
## T- 20 80
## T+ 280 120prop.table(man.th.ov.tab, 1)## overlijden
## therapie O- O+
## T- 0.2 0.8
## T+ 0.7 0.3# Selecteren van vrouwen
d.vrouw <- d[d$geslacht == "vrouw",]
vrouw.th.ov.tab <- table(d.vrouw$therapie, d.vrouw$overlijden, dnn = c("therapie", "overlijden"))
vrouw.th.ov.tab## overlijden
## therapie O- O+
## T- 368 32
## T+ 97 3prop.table(vrouw.th.ov.tab, 1)## overlijden
## therapie O- O+
## T- 0.92 0.08
## T+ 0.97 0.03Met deze data willen we natuurlijk ook RD, RR en OR uitrekenen. Dit kun je doen door gebruik te maken van het pakket EpiR. Zorg dat die geïnstalleerd is voordat je dit commando gaat gebruiken.
# install.packages("epiR")
# Voor mannen
epi.2by2(dat = man.th.ov.tab, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 20 80 100 20.00 (12.67 to 29.18)
## Exposure- 280 120 400 70.00 (65.25 to 74.45)
## Total 300 200 500 60.00 (55.56 to 64.32)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 0.29 (0.19, 0.43)
## Inc odds ratio 0.11 (0.06, 0.18)
## Attrib risk in the exposed * -50.00 (-59.03, -40.97)
## Attrib fraction in the exposed (%) -250.00 (-427.35, -140.88)
## Attrib risk in the population * -10.00 (-16.21, -3.79)
## Attrib fraction in the population (%) -16.67 (-17.44, -15.75)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 83.333 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population unitsKijk eens naar de tabel die gegenereerd is, en de namen die de kolommen en rijen hebben gekregen. Wat valt je op? Zal deze analyse de correcte RR (in deze functie heet die “inc risk ratio”), OR en RD (hier: “Attrib risk in the exposed”) geven?
Het probleem is de volgorde van de kolommen. We willen dat de tabel die gegenereerd wordt een bepaald format heeft, namelijk de mensen met de bijzondere uitkomst (die in deze studie overlijden) in de eerste kolom en de mensen met de bijzonder behandeling (die in deze studie therapie krijgen) in de eerste rij. Alleen dan krijg je de juiste informatie over het risico op overlijden van de groep met therapie ten opzichte van geen therapie. Dit is nu niet het geval. Je zou, als oplossing, een nieuwe uitkomstvariabele kunnen maken waarvan de codering andersom is, maar R kan dit ook voor je hercoderen met de functie factor:
# Hercoderen van overlijden zodat O+ eerst komt
d$overlijden <- factor(d$overlijden, levels = c("O+", "O-"))
# Opnieuw tabellen maken
d.man <- d[d$geslacht == "man",]
man.th.ov.tab <- table(d.man$therapie, d.man$overlijden, dnn = c("therapie", "overlijden"))
man.th.ov.tab## overlijden
## therapie O+ O-
## T- 80 20
## T+ 120 280# Nu de juiste analyse
epi.2by2(dat = man.th.ov.tab, method = "cohort.count", conf.level = 0.95, outcome = "as.columns")## Outcome+ Outcome- Total Inc risk *
## Exposure+ 80 20 100 80.00 (70.82 to 87.33)
## Exposure- 120 280 400 30.00 (25.55 to 34.75)
## Total 200 300 500 40.00 (35.68 to 44.44)
##
## Point estimates and 95% CIs:
## -------------------------------------------------------------------
## Inc risk ratio 2.67 (2.23, 3.19)
## Inc odds ratio 9.33 (5.47, 15.93)
## Attrib risk in the exposed * 50.00 (40.97, 59.03)
## Attrib fraction in the exposed (%) 62.50 (54.98, 68.60)
## Attrib risk in the population * 10.00 (3.79, 16.21)
## Attrib fraction in the population (%) 25.00 (21.80, 28.39)
## -------------------------------------------------------------------
## Uncorrected chi2 test that OR = 1: chi2(1) = 83.333 Pr>chi2 = <0.001
## Fisher exact test that OR = 1: Pr>chi2 = <0.001
## Wald confidence limits
## CI: confidence interval
## * Outcomes per 100 population unitsLet op! Je moet nu natuurlijk wel alle stappen herhalen die je na het “repareren” van de dataset hebt gezet. Als het goed is resulteert dit in de juiste RR / OR die je ook handmatig uit hebt gerekend bij a. van deze vraag.
Als laatste kun je in de output zien dat hier ook de chi-kwadraat toets uitgevoerd wordt op de data. Vergelijk deze met de toets die je bij f. van deze vraag hebt gedaan. Komt hier hetzelfde uit?
Uiteraard kun je bovenstaande analyses herhalen voor vrouwen.